Not long ago I wrote about one chapter of my PhD thesis that was published and this week I had another paper published in Molecular Ecology. We first submitted it in spring of 2020 and got reviews back after a few weeks. Reviewers asked for a lot of revisions and suggested we perform different analyses to those we had presented in some parts. All-in-all, it took us a year to make changes, additional analyses, and finally submitting a revised version of the manuscript in April of this year. Completing a project from data collection to publication is often a bit of a test of endurance.
This paper was part of a long-standing collaboration between my PhD supervisor, Professor Mike Ritchie, and Finnish colleagues from the University of Jyväskyla: Dr. Maaria Kankare and Professor Anneli Hoikkala. Mike, Maaria, and Anneli have worked for several years to understand the evolution of cold-tolerance and population divergence in Drosophila montana.
D. montana is an extremely cold-tolerant species of Drosophila and can occur at very high latitudes throughout the northern hemisphere. It also exhibits an adaptive overwintering behaviour whereby females that have emerged late in the year, instead of reproducing, enter an overwintering state called ‘diapause’. They then emerge again in spring to reproduce. Populations of D. montana also exhibit a certain extent of reproductive isolation, a crucial stage of speciation whereby individuals from diverging populations of the same species start to have trouble producing offspring when mating with individuals from another population. This makes the species an ideal model system for the study speciation. Other work on this species has shown, for example, that females tend to prefer the songs of males from their population, and that crosses between individuals from different populations produce fewer eggs and offspring than crosses between individuals from the same population.
In our study we wanted to identify regions of the genome where genetic differences between populations was associated with ecological differences between populations. To do this we wanted information about genetic markers throughout the genome from several populations that also differed in ecology. Here it might be good to have a short diversion on genetic markers and why they are of interest. When we perform whole-genome sequencing to compare populations, we are primarily concerned with SNPs, that is Single Nucleotide Polymorphisms. This is a technical term for locations in the genome where individuals within a population differ in the DNA base. In the figure below, for example, there is a small cartoon of 10 locations in the genome as seen in 5 different individuals from one population. At one of the locations, 2 individuals have an A (in red), and 3 individuals have a C (in blue), this site is therefore said to be “polymorphic” and because the site represents a single nucleotide, it becomes a “single nucleotide polymorphism”, or SNP (pronounced “snip”). We can also say that the frequency of the “A” allele is 2/5 (or 0.4). This on it’s own doesn’t tell us very much but when we sequence many individuals and many populations we can begin to see patterns. For example, we can find sites in the genome where the frequency of a particular allele has a relationship with some climatic variable, see the second figure below. In this way we can start to say interesting things about whether natural selection might be involved in determining the allele frequencies.
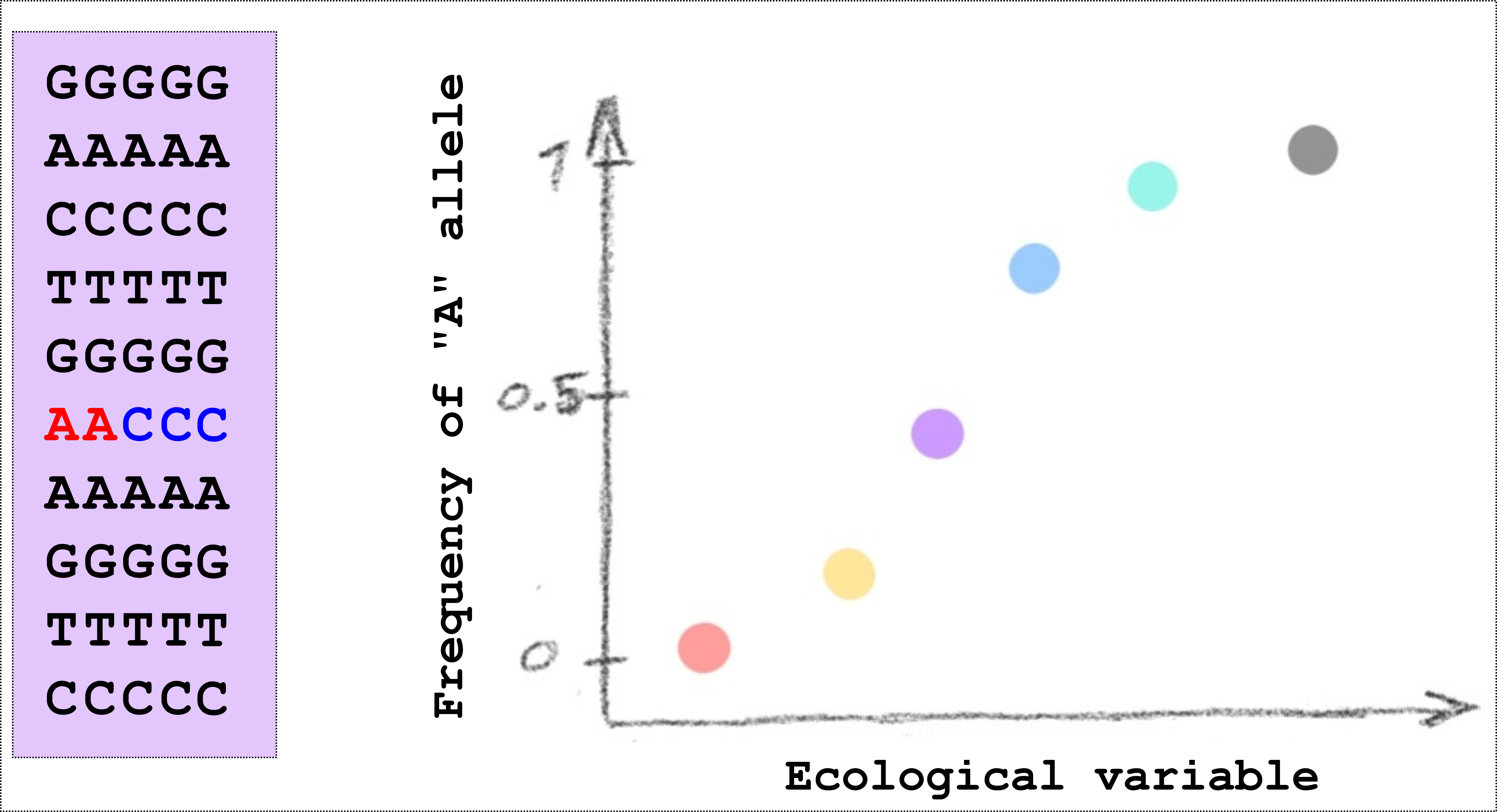 |
|---|
| A small cartoon illustrating a Single Nucleotide Polymorphism (SNP). An illustration of a SNP where the allele frequency in different populations (shown as different coloured points) is related to some ecological variable. |
We investigated 24 populations (see the figure below) to get a sense for climatic and seasonal variation across them. We then generated genome-sequencing data from 6 of these populations for which we also conducted standardised experiments to determine how cold-tolerant they are. We now had ecological variables that represented local climate and seasonality as well as phenotypic data that represented the degree of cold-tolerance in each of 6 populations. We also had genome-sequencing data that gave data on how common specific alleles at ~2 million genetic markers in each population. We then identified genetic markers where the allele frequency differences between populations was associated with ecological and/or phenotypic differences between populations. Importantly, the method we used could account for similarities between populations that were due to them sharing a common ancestor. We need to be able to tell the difference between populations being genetically similar due to similar ecology and populations being genetically similar simply because they are related.
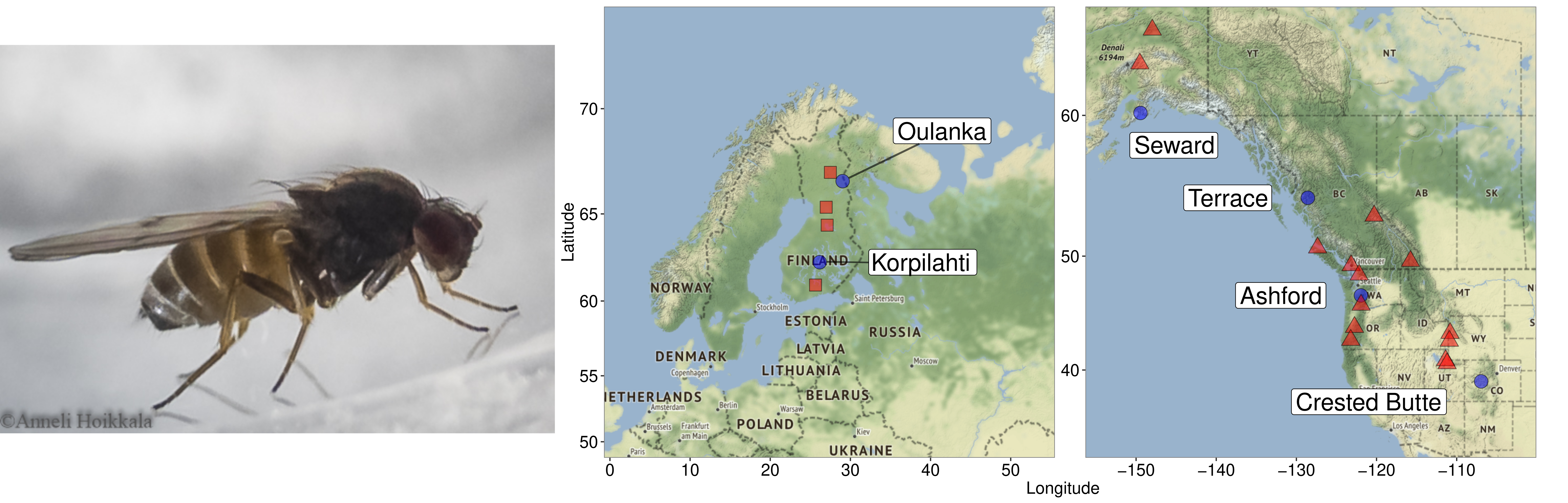 |
|---|
| A female D. montana (credit to Anneli Hoikkala for the image). Shown in the maps are locations of the populations we sampled. Populations in red were used for genetic analyses |
Depending on the ecological/phenotypic variable we used, we identified between 3,468 and 8,468 markers with a strong association in allele frequencies. Indicating that these variables were responsible for the allele frequency differences between populations. We could then identify which genes were near these genetic markers as well as the biochemical or metabolic processes that these genes are involved in. It turns out that we get very similar results when using either the climatic data or the phenotypic data. This suggests that the differences in climate/seasonality between populations is really responsible for the differences in phenotypes between populations, and that both are associated with the same genetic differences. In other words, differences in climate selects for particular phenotypes, which results in changes in allele frequencies across populations. Evolution by natural selection in a nutshell!
A really cool part of this paper for me is that we were able to link the differences between popoulations quite robustly to results from previous studies in two important ways. First, genes and metabolic processes that have been implicated in being part of the underlying mechanism of cold-tolerance and diapause behaviour from previous studies were also shown to be within the regions of the genome that showed strong differences between populations related to local climatic conditions in our study. To me, this is a strong indication that ecological differences between populations is driving changes in genes that determine these phenotypes. Second, many of these same genes and processes have been shown to be important in setting D. montana apart from other Drosophila species. This suggests that the same process that continues to drive divergence across populations within the species, namely adaptation in response to local ecological conditions, has also driven the divergence of D. montana from other closely related species. This result is a really nice way-marker on the continuum between genetic differences between populations and the origin of new species. The results of many years of work on this species is painting a picture of the very process of biological evolution by natural selection.
The work is not done, of course, there are several other interesting results in the paper that will require more work by us to understand. We’re not putting anyone out of a job just yet.
My co-authors in on this paper were along with a former lab-mate of mine Venera Tyukmaeva, Anneli Hoikkala, Mike Ritchie, and Maaria Kankare. The full paper is open access, and you can read the paper here if you want to learn more about this. Once again, please get in touch if you have questions!